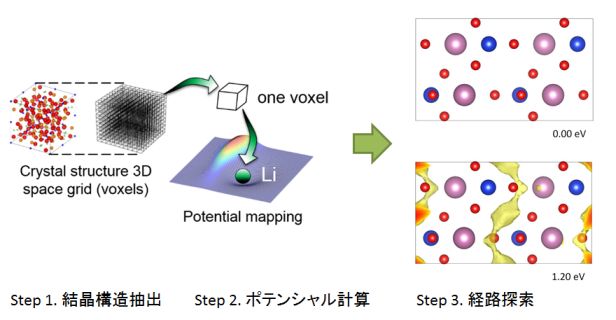
【リチウムイオン電池用電極・固体電解質材料】
★例 第一原理分子動力学法によるガーネット型固体電解質材料のイオン伝導機構解析(参考文献 Link1)
ガーネット型酸化物は高いリチウムイオン導電性かつ耐リチウム金属安定性を示すことから、実用材料として注目されている。本研究では、第一原理分子動力学計算を行うことでイオン導電性を評価し、実験値に適合する導電特性を再現することに成功した。この結果について、原子レベルの観点でイオンの伝導機構を調査したところ、2つ以上のリチウムイオンのが協奏的にホッピングする機構や、リチウムイオンの占有するサイトの詳細情報を得ることができた。
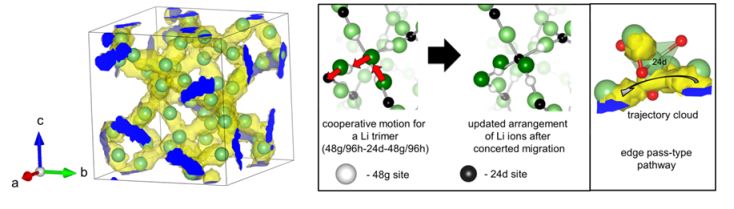
図: 左) リチウムイオン伝導パスを可視化したもの 右)リチウムイオンの協奏的ホッピングメカニズム
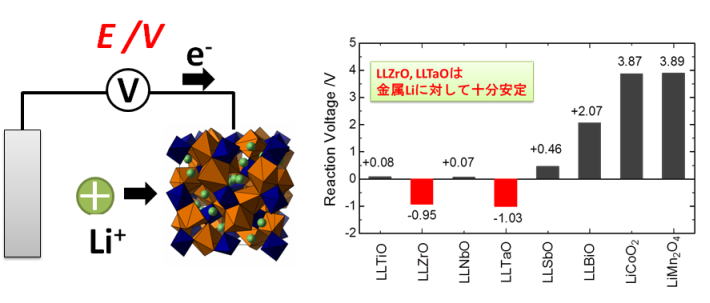
★例 リチウムイオン導電性ガーネット型酸化物における金属リチウムに対する安定性 (参考文献 Link1)
ガーネット型酸化物は高いリチウムイオン導電性かつ耐リチウム金属安定性を示すことから、実用材料として注目されている。本研究では、第一原理計算と実験を組み合わせて、様々な組成と構造における相安定性を調査し、金属リチウムに対する安定性を確保するための組成と構造に関する材料設計の指針を明らかにした。
 ★例 Nudged-Elastic-Band法(NEB法)による正極材料のイオン伝導機構解析(参考文献 Link1, Link2)
★例 Nudged-Elastic-Band法(NEB法)による正極材料のイオン伝導機構解析(参考文献 Link1, Link2)
スピネル型LiMn2O4は安価なリチウム電池の電極材料として注目されている。この材料では、Mnの一部を別の遷移金属で置換すると出力特性が向上すると考えられている。本研究では第一原理計算を用いて、その機構がMnの電子構造に深くかかわっていることを明らかにした。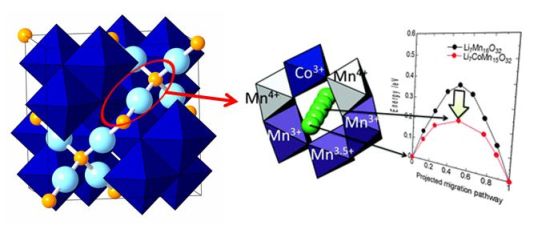
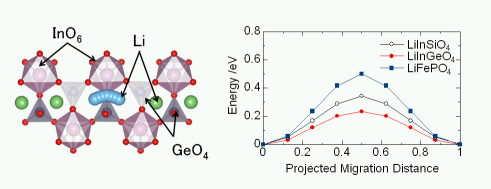
★例 Nudged-Elastic-Band法(NEB法)による新規リチウムイオン伝導体の設計(参考文献 Link1, Link2, Link3)
一般に第一原理バンド計算では、基底状態(絶対零度で最安定なエネルギー状態)の計算を取り扱うため、イオンのホッピング伝導のように励起状態を扱うことは難しい。しかし、近年ではサイト間ホッピングするイオン種の座標や加わる力に拘束条件を与えることで、ホッピングの様子を再現することができるようになってきた(NEB法など)。われわれはNEB法を適用することにって、新しいLiイオン導電体であるオリビン型LiInGeO4, LiInSiO4を提案した。
★例 ペロブスカイト型リチウムイオン伝導性材料の欠陥配列 (参考文献 Link1)
ペロブスカイト型(Li, La)TiO3材料は酸化物として最大のリチウムイオン導電性を有していることで知られている。この材料はペロブスカイトAサイトに分布するLiイオンが伝導すると考えられているが、AサイトにはLaと空孔も同居しており、その配置自由度が膨大であるため(Li/La/空孔の並び方が何通りも考えられるということ)からシミュレーションをするのが難しかった。われわれは、乱数を用いたモンテカルロ法のポテンシャルパラメーターを第一原理バンド計算によって決定する(クラスター・イクスパンジョン法)ことで、非経験パラメーターに基づくモンテカルロ・シミュレーションを実施し、確度の高いLa/空孔配列を見出た。更に、重要なLa/空孔配列におけるリチウムイオン拡散を原子レベルで再現することに成功した。
 ★例 古典力場を利用したハイスループット・全自動リチウムイオン導電能評価 (参考文献 2015)
★例 古典力場を利用したハイスループット・全自動リチウムイオン導電能評価 (参考文献 2015)
セラミックス中のイオン導電性評価にはNEB法や分子動力学法が使われているが、これらの計算法は入力の手間と計算時間が膨大なため、探索空間を広げた数千件程度の材料に対する評価には不向きであった。この研究では、古典力場と浸透理論を活用することで、イオン導電性評価の高速化と全自動化の方法を提案することができた。