今日、第一原理計算等に代表される計算科学的アプローチは、機能性材料の組成・構造・物性の関係を解明する重要なツールとなっている。しかし、解析には適していても新しい材料を探索することは実は難しい。これは、「組成・構造」から「物性」の解析はできても、「物性」から望ましい「組成・構造」の探査は容易にできないからである。したがって計算者は事前に十分な候補材料選定や計算計画を練ってから研究を行わなければならず、成功のためには多分に「運」の要素も加味しなければならない。つまり実験的アプローチとなんら変わらない状況になっている。
我々は、優れた機能を有する材料を計算科学的に探査するために、計算材料学のアプローチに情報学の概念を付与した「マテリアルズ・インフォマティクス」の考え方を導入し、そのモデル的研究を行っている。
また従来の計算では、初期構造を与えなければ計算することができず、既知構造の材料しか計算対象となりえなかったが、Oganovらが開発した進化的アルゴリズムなどにより構造を第一原理的に予測することも可能になってきた。我々はこのような新しいアルゴリズムを用いて新たな視点から電池材料の特性評価を行ったり、新材料の発見を目指している。
★例 オリビン型結晶構造を持つリチウムイオン導電性LiMXO4材料の最適組成探査 (参考文献 2012, 2014)
我々は典型元素から構成されるオリビン型結晶構造がリチウムイオン導電性に優れることを以前報告した[*]。この材料について、考えられる組成の組み合わせは定比のものだけでも約70種類に及ぶ。このような広範な組成について材料を第一原理計算でイオン導電性を評価することは計算時間的に困難であることから、本研究では約10サンプルの第一原理計算結果を情報学的に学習し、のこり50種類の組成組み合わせについて多変量解析によって一気に導電性を評価する方法を提案した。多変量解析には、PLS法(link1)やニューラルネットワーク法(link2)などを適用している。
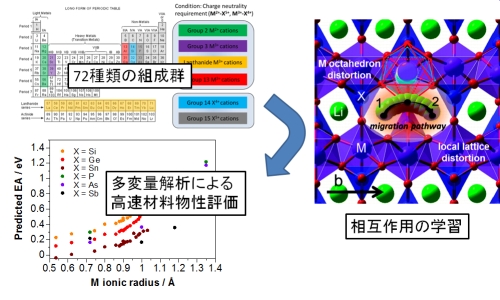
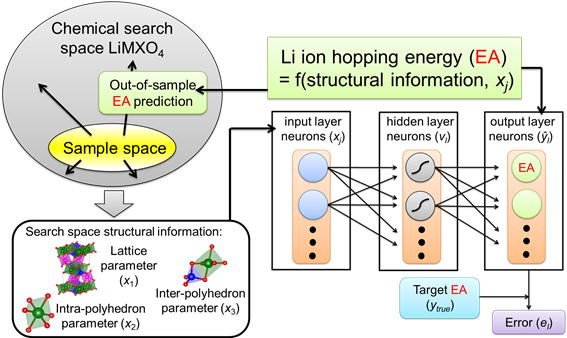 ▲ ニューラルネットワーク回帰分析による計算手順
▲ ニューラルネットワーク回帰分析による計算手順
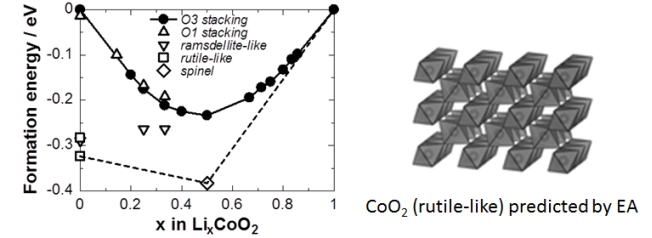
★例 進化的アルゴリズムを用いたLixCoO2正極の第一原理構造予測 (参考文献 2012)
リチウムイオン電池を充電状態のままにすると、正極の遷移金属は酸化数の大きな不安定状態になり、長期的には構造が変形し劣化するものと考えられる。このような構造変化を実験的に評価することは膨大な時間を必要とするため困難である。一方、本研究では進化的アルゴリズムを使うことで最安定相の構造を第一原理的に調査し、充電相の安定性について議論した。(進化的アルゴリズムにはUSPEXを使用)