2020年以降のページは、以下からアクセスしてください
1)材料シミュレーション
2)材料インフォマティクス
■ 計算化学によるセラミックス材料設計
第一原理バンド計算やモンテカルロ計算などの先端計算手法を用いて、機能性セラミックス材料の結晶構造や電子構造と材料機能の関係を解析し、よりすぐれた新たな材料を設計する指針を得ることを目指しています。主には「リチウムイオン電池材料」や「燃料電池・酸素透過膜材料」のようなエネルギー変換セラミックス材料をターゲットとして研究を行っています。
(1) イオン導電性材料の設計
(2) マテリアルズ・インフォマティクス
(3) 誘電体材料の物性予測
(4) 第一原理熱力学
(5) セラミックス材料の強相関系電子構造と物性
(6) X線吸収スペクトルの理論計算
(7) 表面・界面化学反応の第一原理計算研究
リチウムイオンや酸化物イオンの固体内拡散は、リチウムイオン電池や燃料電池などの電気化学デバイスの特性・性能を決定する重要な現象である。高いイオン拡散能をもった材料を設計するためには、欠陥生成エネルギーの推定やサイト間ホッピングにおけるポテンシャル障壁の見積もりが必要である。
研究例:第一原理分子動力学法による固体電解質材料のイオン伝導機構解析
ガーネット型酸化物は高いリチウムイオン導電性かつ耐リチウム金属安定性を示すことから、実用材料として注目されている。本研究では、第一原理分子動力学計算を行うことでイオン導電性を評価し、実験値に適合する導電特性を再現することに成功した。この結果について、原子レベルの観点でイオンの伝導機構を調査したところ、2つ以上のリチウムイオンのが協奏的にホッピングする機構や、リチウムイオンの占有するサイトの詳細情報を得ることができた。ガーネットのほかにも固体電解質材料候補に対する第一原理分子動力学法計算の研究を行っている。
【参考文献例】 Garnet型材料: 2013, 2015 NASICON型材料: 2016, 2017, 2018, 2019
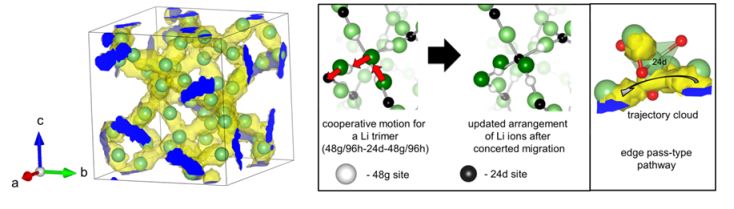
図 ガーネット型固体電解質材料の研究成果例(伝導パスと協奏的ホッピング現象)
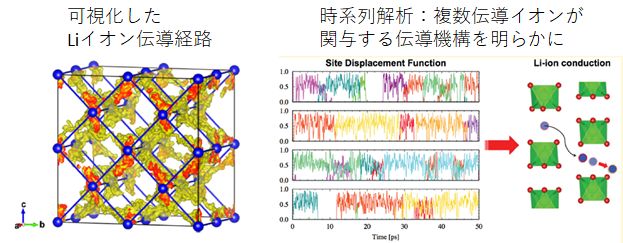
図 NASICON型 LiZr2(PO4)3材料におけるLiイオンの拡散経路
研究例:固体電解質の電位窓:正極・負極に対する安定性
ガーネット型酸化物は高いリチウムイオン導電性かつ耐リチウム金属安定性を示すことから、実用材料として注目されている。本研究では、第一原理計算と実験を組み合わせて、様々な組成と構造における相安定性を調査した。負極としては金属リチウムに対する安定性を確保するための組成と構造に関する材料設計の指針を抽出した。一方正極との安定性については、電極コンポジットに含まれるカーボンとの反応によって酸化分解が発生することを明らかにした。どちらも実験及び計算の観点から機構を解明した。
また、材料の熱力学的安定性は凸包計算により評価できることから、数百~数万の第一原理計算データベースと連携して、電位窓の網羅評価や複雑な分解反応の解析なども実験と連携するなどして行っている。
【参考文献例】 ガーネット型材料:2012, 2016 NASICON型材料:2017 データベースとの連携など:2019, 2020(日本語)
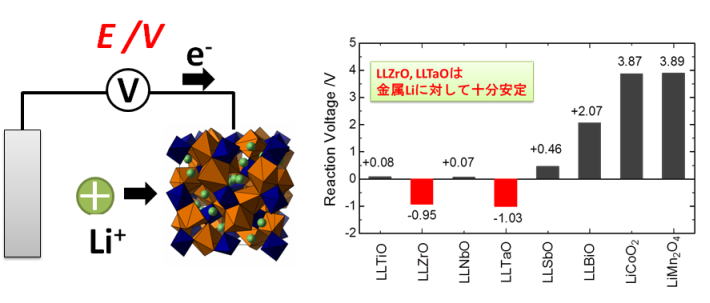
図 負極(Li金属)との反応性評価
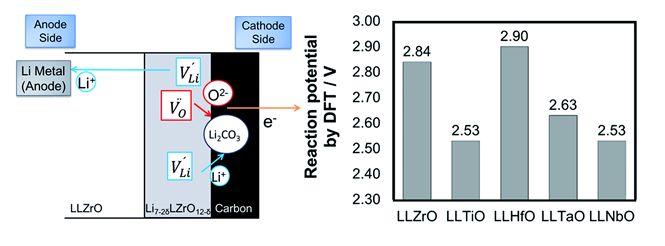
図 正極コンポジット中のカーボンとガーネット材料の充電反応による分解反応
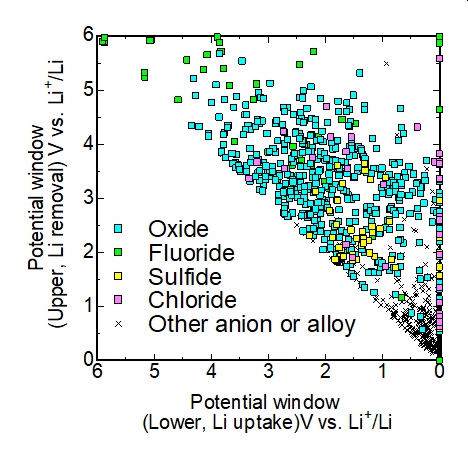
図 凸包計算によって網羅的に評価されたリチウムイオン電池固体電解質材料の電位窓 (縦軸:正極に対する耐酸化性、横軸:負極に対する耐還元性)
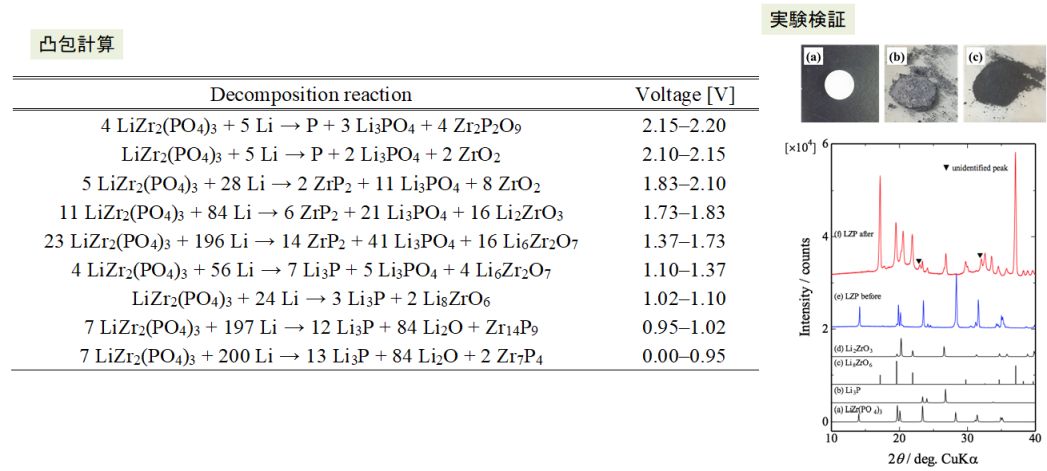
図 凸包計算で詳細解析されたNASICON型LiZr2(PO4)3固体電解質材料の負極に対する還元分解反応の電位依存性解析結果(実験との対比)
★例 Nudged-Elastic-Band法(NEB法)による正極材料・固体電解質材料などのイオン伝導機構解析(参考文献 2012, 2014, 2019 など)
蓄電池の電極材料や固体電解質材料におけるリチウムイオン伝導機構の解明は、合理的な材料設計に結び付けられる。例えば、イオンのホッピングの瞬間は、実験的な解析が極めて困難である。NEB法によりホッピングの瞬間の周辺構造の歪構造の変化や、電子構造の変化(周辺イオンのイオン価数の瞬間的変化)などを解析している。また、固体電解質材料の計算結果などをデータとして蓄積し、マテリアルズ・インフォマティクスなどの手法開発にも利用している。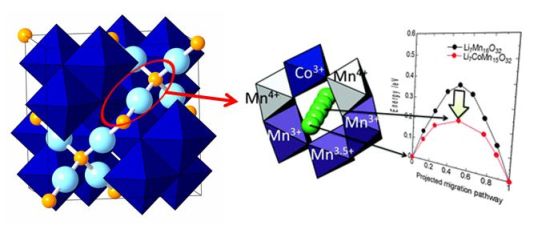
▲スピネル型LiMn2O4材料に対してNEB法を用いてLiイオンホッピングを解析した例。電子構造をDFT+U法で計算すると、Liホッピングの瞬間に周辺のMnイオンの価数が変動することが明らかとなった。これによりCo置換などの優位性などを解明した。
★例 リチウムイオン伝導性材料における欠陥配列 (参考文献 2009, 2020)
ペロブスカイト型(Li, La)TiO3材料は酸化物として最大のリチウムイオン導電性を有していることで知られている。この材料はペロブスカイトAサイトに分布するLiイオンが伝導すると考えられているが、AサイトにはLaと空孔も同居しており、Li/La/空孔の並び方が無数に考えられることから、シミュレーションをするのが難しい。そこで、高精度第一原理計算の結果を利用してモンテカルロ法を適用し(クラスター展開法/Cluster Expansion法)、イオンの配列を見出せた。また類似のLa2/3NbO3材料では、複雑な透過電子顕微鏡回折像も再現することにも成功している。
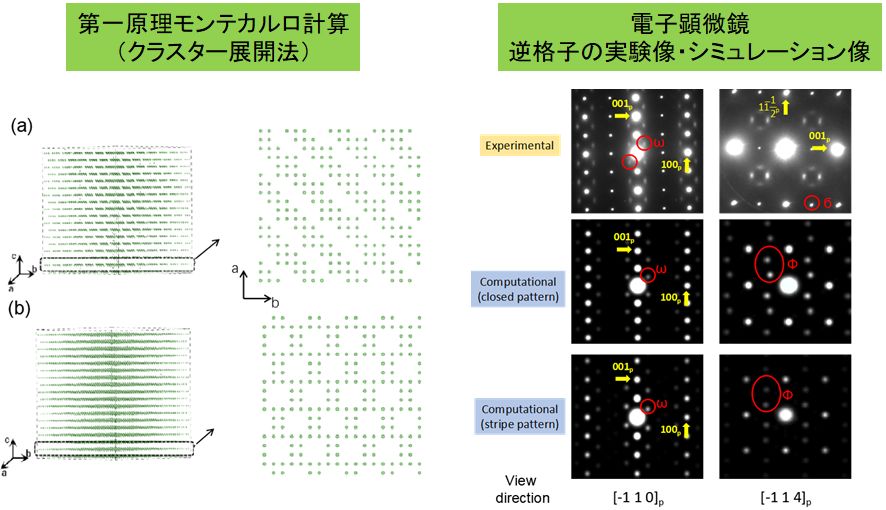
▲リチウムイオン導電性La2/3NbO3材料について第一原理モンテカルロ計算を行った結果。2つの特徴的な長周期配列が確認され、実験的得られた逆格子像と一致した。
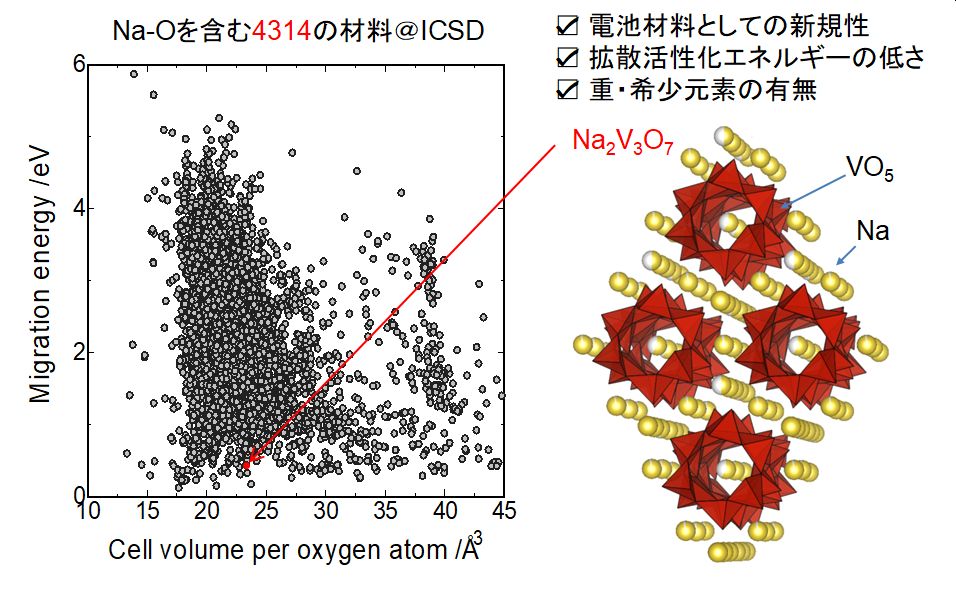
★例 古典力場を利用したハイスループット・全自動リチウムイオン導電能評価 (参考文献 2015, 2018, 2020)
セラミックス中のイオン導電性評価にはNEB法や分子動力学法が使われているが、これらの計算法は入力の手間と計算時間が膨大なため、探索空間を広げた数千件程度の材料に対する評価には不向きであった。この研究では、古典力場と浸透理論を活用することで、イオン導電性評価の高速化と全自動化の方法を提案することができた。
この方法論を活用して、実際にナトリウムイオン電池やマグネシウムイオン電池の新材料を実験的に発見する成果に結びつけた。また、大量に得られたデータをマテリアルズ・インフォマティクス研究の試験データとして活用している(後述)。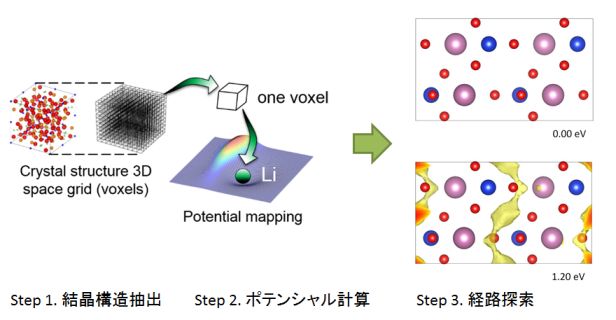
▲ 古典力場計算+浸透理論を組み合わせた全自動イオン導電性評価法
▲数千件のデータベースに掲載された材料候補に対して網羅計算を実施。電池材料候補を抽出し実験的に性能を立証することに成功した。
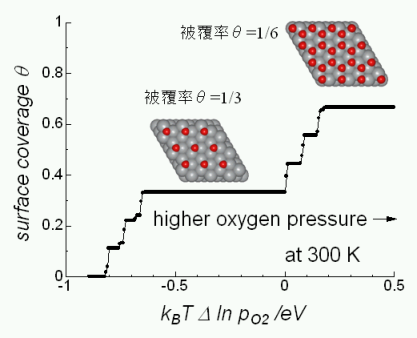
★例 還元セリア(CeO2-d)における電子・イオン混合伝導 (参考文献 2012)
酸化物イオンと電子混合導電性を示す酸素透過膜は水素製造・二酸化炭素固定化のカギとなる材料である。われわれは第一原理計算によってイオンと電子の間に強い相互作用が働いており、イオンと電子が協奏的にサイト間ホッピングすることで活性化エネルギーも低下することが明らかとなった。
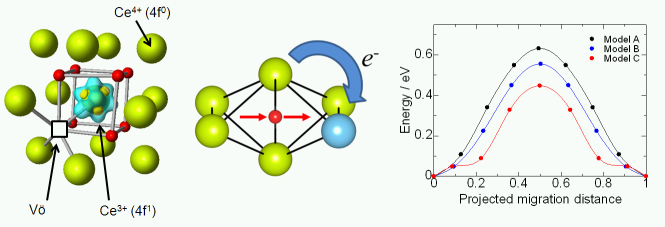
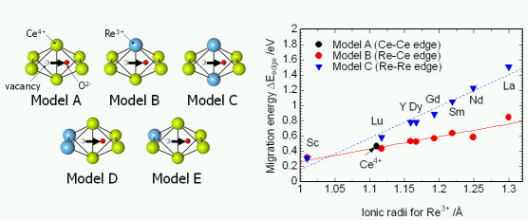
★例 希土類置換セリア(Re-doped CeO2)における酸化物イオン伝導 (参考文献 2009)
希土類置換セリア(Re-doped CeO2)は、600℃程度の中温域でも高い酸化物イオン伝導能をもつことから、固体酸化物型燃料電池の固体電解質材料などに応用されることが期待されている。われわれは第一原理計算によって酸素空孔=希土類イオン会合エネルギー(上図)と酸化物イオンのホッピングのエネルギー障壁エネルギー(NEB法)を算出した。ドープする希土類イオンのイオン半径に対して、それぞれのエネルギーに対応関係があることを示した。更に、統計力学的な取り扱いによって、希土類イオンドープ量に最適値があることや、アレニウスプロットが屈曲することなどを再現できた。
★例 SOFC空気極用ペロブスカイト型酸化物GBCOにおける相転移反応 (参考文献 2013)
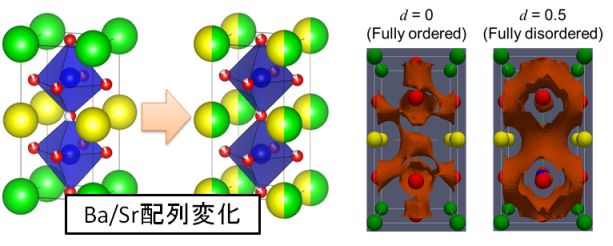
GdBaCo2O5+d (GBCO)はSOFC空気極材料や酸素透過膜材料として注目されている。この材料は、高温で相転移を示す。本研究では、第一原理計算とモンテカルロ法を併用することで酸素・空孔配列の秩序/無秩序相転移を解明した。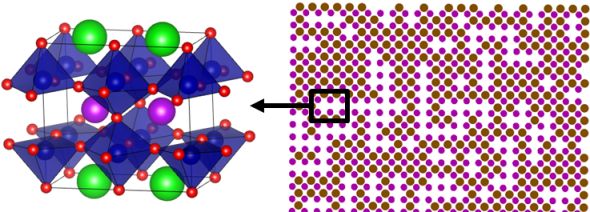
★例 SOFC空気極用ペロブスカイト型酸化物BSCFの酸素輸送能 (参考文献 2013)
ペロブスカイト型BSCFはSOFC空気極材料や酸素透過膜材料として注目されている。より優れた酸素輸送能を得るために、カチオン配列に注目し、分子動力学計算によって、拡散係数の比較を行った。
(2) マテリアルズ・インフォマティクス
今日、第一原理計算等に代表される計算科学的アプローチは、機能性材料の組成・構造・物性の関係を解明する重要なツールとなっている。しかし、解析には適していても新しい材料を探索することは実は難しい。これは、「組成・構造」から「物性」の解析はできても、「物性」から望ましい「組成・構造」の探査は容易にできないからである。したがって計算者は事前に十分な候補材料選定や計算計画を練ってから研究を行わなければならず、成功のためには多分に「運」の要素も加味しなければならない。つまり実験的アプローチとなんら変わらない状況になっている。
我々は、優れた機能を有する材料を計算科学的に探査するために、計算材料学のアプローチに情報学の概念を付与した「マテリアルズ・インフォマティクス」の考え方を導入し、そのモデル的研究を行っている。
また従来の計算では、初期構造を与えなければ計算することができず、既知構造の材料しか計算対象となりえなかったが、Oganovらが開発した進化的アルゴリズムなどにより構造を第一原理的に予測することも可能になってきた。我々はこのような新しいアルゴリズムを用いて新たな視点から電池材料の特性評価を行ったり、新材料の発見を目指している。
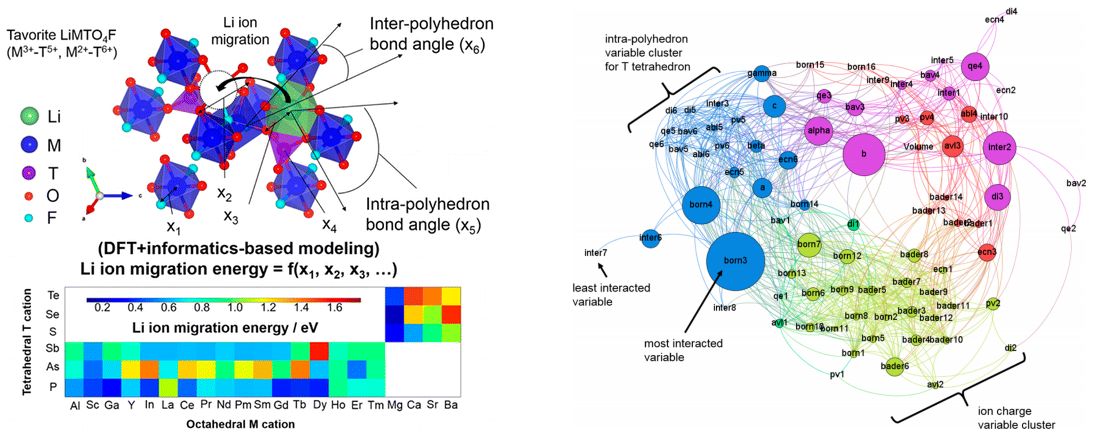
★例 オリビン型やタボライト型結晶構造を持つリチウムイオン導電性材料の最適組成探査 (参考文献 2012, 2014, 2015)
我々は典型元素から構成されるオリビン型結晶構造がリチウムイオン導電性に優れることを以前報告した。この材料について、考えられる組成の組み合わせは定比のものだけでも約70種類に及ぶ。このような広範な組成について材料を第一原理計算でイオン導電性を評価することは計算時間的に困難であることから、本研究では約10サンプルの第一原理計算結果を情報学的に学習し、のこり50種類の組成組み合わせについて多変量解析によって一気に導電性を評価する方法を提案した。多変量解析には、PLS法やニューラルネットワーク法などを適用している。現在はこの方法論を応用して、その他の材料系にも応用している。
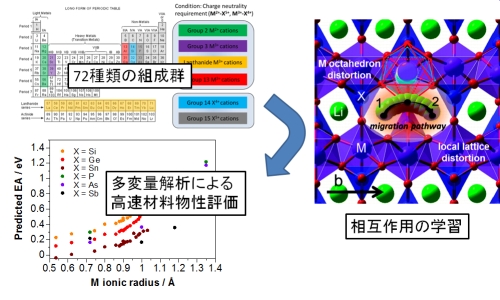
▲ 多変量解析による材料物性予測
▲タボライト構造における記述子定義と記述子間の相関関係
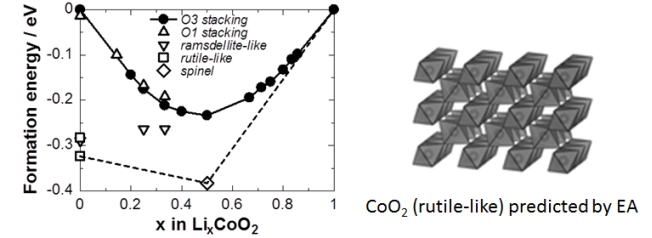
★例 進化的アルゴリズムを用いたLixCoO2正極の第一原理構造予測 (参考文献 2012)
リチウムイオン電池を充電状態のままにすると、正極の遷移金属は酸化数の大きな不安定状態になり、長期的には構造が変形し劣化するものと考えられる。このような構造変化を実験的に評価することは膨大な時間を必要とするため困難である。一方、本研究では進化的アルゴリズムを使うことで最安定相の構造を第一原理的に調査し、充電相の安定性について議論した。(進化的アルゴリズムにはUSPEXを使用)
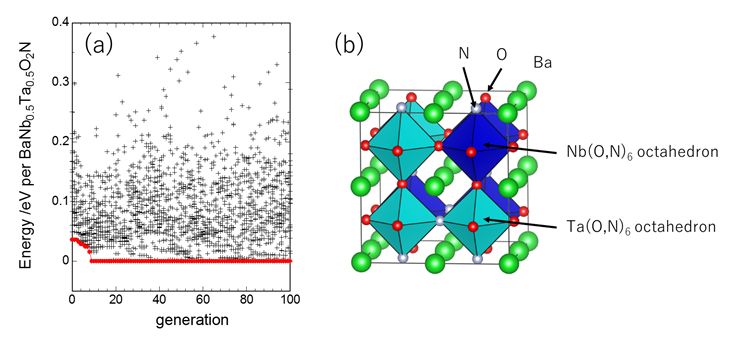
★例 遺伝的アルゴリズムと第一原理計算を利用した複雑組成のイオン配列決定 (参考文献 2016)
光触媒材料であるBaNbO2N-BaTaO2N材料では、Nb/Ta, O/Nのイオンの配列自由度による複雑性のため計算用の構造決定が容易ではない。本研究では独自に作成した遺伝的アルゴリズムのコードを用いて、効率的に最安定構造を決定し、各種の光触媒特性について考察した。(信州大学との共同研究)
★例 汎用記述子の開発(参考文献 2018)
結晶構造データベースを利用したマテリアルズ・インフォマティクスでは、組成や結晶構造を数値化(記述子という)する必要がある。われわれはヒストグラム表示を利用することで、任意の組成・結晶構造を記述子に変換することを提案し、さらに有効に材料物性を予測することを確認した。
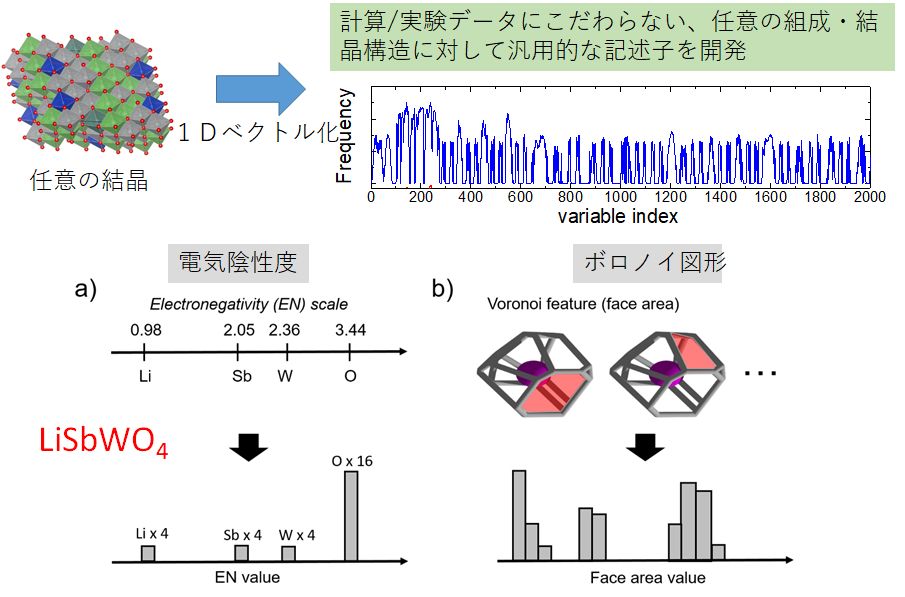
▲ 汎用性の高いヒストグラム記述子の概要
★例 複数の組成・構造を有する材料データベースのイオン導電性物性(固体電解質特性)予測(参考文献 2019, 2020)
既存の結晶構造データベース(ICSDなど)から全固体電池用固体電解質を探索するには、データベースのボリュームから考えると数百~数万結晶構造データに対する評価が必要である。このような課題を解決できるインフォマティクスアプローチを検証するため、記述子にヒストグラム形式を用いた汎用記述子、イオン電導物性に力場計算で簡易的・網羅的に求めた結果を用いスキームの妥当性を評価した。その結果、回帰分析による予測はある程度の効果は認められるものの、要求する予測能力は発揮できないことが分かった。一方、ベイズ最適化を用いることで、全体の1/10の計算労力でほぼ最適材料(最もイオン導電性の高い材料)を発見できることが分かった。
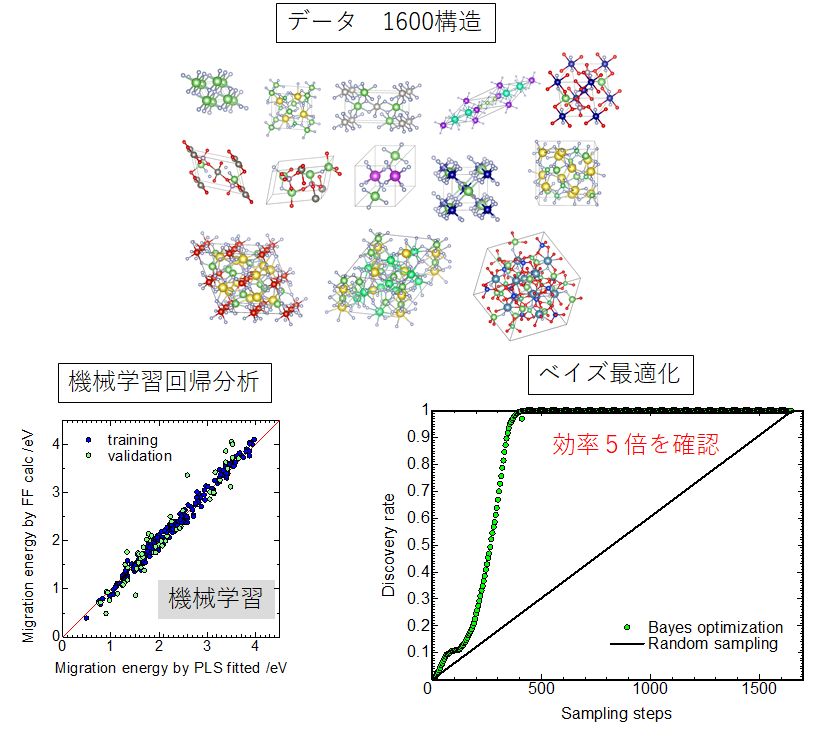
▲構造について束縛のない条件で1600構造を抽出。得られた結果を汎用ヒストグラム記述子に変換し、回帰分析やベイズ最適化を実施した結果。
★ベイズ最適化を用いたコドープ組成最適化 (参考文献 2020)
固体電解質母材料に不純物イオンを導入すると、イオン導電性が劇的に向上することがあり、有効な方法として知られている。しかし不純物イオンの導入パターンは膨大となり、材料組成最適化には膨大な時間とコストを費やすことが多い。本研究では材料シミュレーションで導出したイオン導電性データに対して、ベイズ最適化(BO)により最適組成を得るまでに必要な計算量を見積もっており、BOが非常に有効な手法であることを明らかにした。
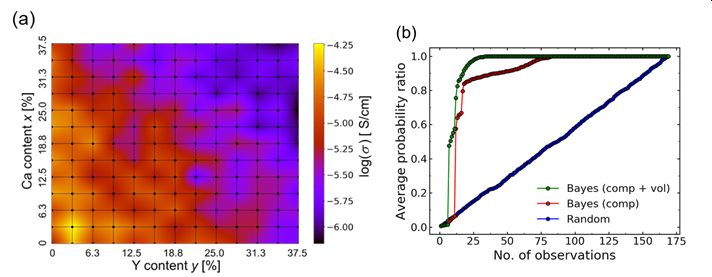
▲構造について束縛のない条件で1600構造を抽出。得られた結果を汎用ヒストグラム記述子に変換し、回帰分析やベイズ最適化を実施した結果。
誘電体や圧電・強誘電特性を有する材料は、エレクトロニクスを支える基盤材料である。従来、誘電特性は材料が純粋イオン結晶であるという視点から理解されてきたが、より定量的には分極に伴う動的な電子構造変化も重要であることが知られるようになってきた。われわれは、DFPT (Density Functional Perturbation Theory)を用いて、Born電荷などを導出し定量的な材料評価を実践している。
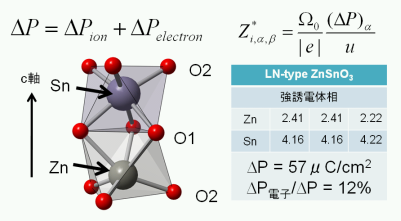
★例 LiNbO3型結晶構造を持つZnSnO3材料の誘電特性1 (参考文献 2010, 2014)
最近、高圧合成法によりLiNbO3型結晶構造を持った極性酸化物ZnSnO3材料が合成された。この新材料の強誘電特性やBorn電荷を算出し、LiNbO3型材料の計算結果と比較考察を行った。(学習院大学との共同研究)
(4) 第一原理熱力学
一般的な第一原理バンド計算では、絶対零度、無加圧下の材料の様子しか計算できないため、材料機能と深く関連する有限温度・圧力下での相転移現象などを予測することが難しかった。われわれは、配置のエントロピー、振動のエンタルピーとエントロピーの計算、また機械的特性での材料の電子エネルギーを第一原理計算から導き統計力学理論などと結びつけることで、有限温度や有限圧力下での材料の相安定性について定量的視点から解析することを実践している。
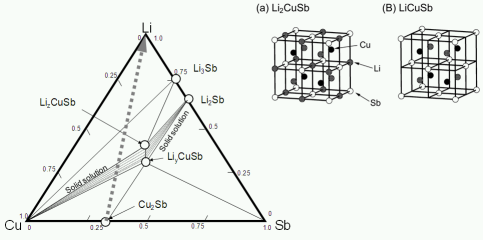
★例 Li-Cu-Sb系合金の三元系相図作成 (参考文献 2007, 2008)
合金系負極材料の電池反応を理解するためには、多元系になるほど複雑な相平衡関係を理解する必要がある。しかし、電池反応中間体は非常に外気に対して不安定であるため、従来のX線回折法などを用いて相図を製作するのは困難である。そこで、われわれは三元系Li-Cu-Sb系合金材料の相図を第一原理計算によって作成し、更に安定したサイクル特性を示すLiCuSb-Li2CuSb-Li3Sb構造の相関係を明らかにした。
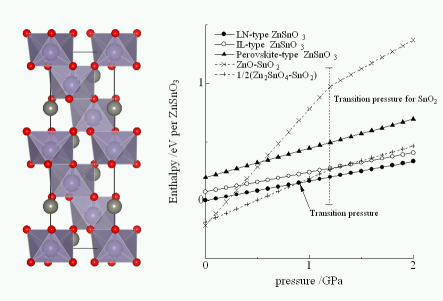
★例 LiNbO3型結晶構造を持つZnSnO3材料の誘電特性2 (参考文献 2010)
Zn2+-Sn4+-O2-三元系材料について、それぞれの相の機械的特性を調べることで、圧力に対する材料の電子エネルギー変化を調べた。この結果、最近報告された極性酸化物材料ZnSnO3が高圧で安定になることを明らかにした。
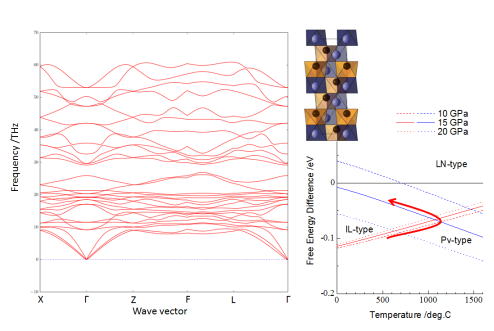
★例 LiNbO3型結晶構造を持つZnTiO3材料の誘電特性 (参考文献 2014)
新たに高圧反応によって合成された新規誘電体材料ZnTiO3の相安定性を、フォノン(格子振動)を考慮した第一原理計算を適用することで決定し、ZnTiO3の合成法を提案しました。(学習院大学との共同研究)
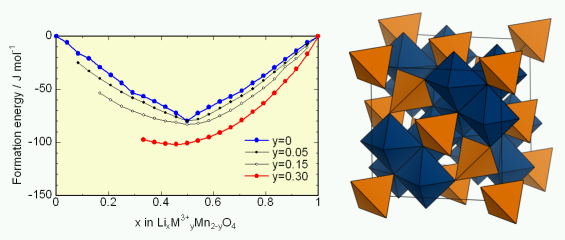
★例 スピネル型系材料の相平衡と反応エントロピー(参考文献 <LiMn2O4a href=”http://pubs.acs.org/doi/abs/10.1021/jp056334y”>2006)
スピネル型LiMn2O4はリチウムイオン電池正極材料として知られているが、充放電反応をするといくつかの相転移が現れる。このような相転移は充放電劣化と深く関係していると考えられている。原子レベルでの相転移メカニズムに関する詳細な知見を得るために、反応エントロピー測定を実施するとともに、実験パラメーターを用いないモンテカルロ計算(クーロン相互作用を使用)を実施した。その結果、リチウムと空孔の規則配列が相転移の際に発生しており、この規則配列は不純物ドープをすることで抑制できることを示した。
(5) セラミックス材料の強相関系電子構造と物性
セラミックス材料のうち、遷移金属酸化物は強相関電子系と呼ばれるものが多い。強相関電子系とは、電子間のクーロン相互作用が強く、結果的に電子が特定の軌道に局在化する。このような系では、密度汎関数理論(DFT)によるバンド計算の結果が実験結果と一致しなくなる傾向があることが知られている。われわれは、DFT+U法やハイブリッド関数(HSE)などを用いて、高い予測精度を維持しつつ、電子伝導(ポーラロンホッピング)や光触媒特性などの物性調査に役立てている。
★例 固体内酸素レドックス反応の第一原理計算による解析
リチウムイオン電池の次世代正極材料として、固体内酸素レドックス反応ができず材料が注目されている。われわれは、酸素周辺の配位構造に注目して、レドックス反応過程を詳細に分析している。
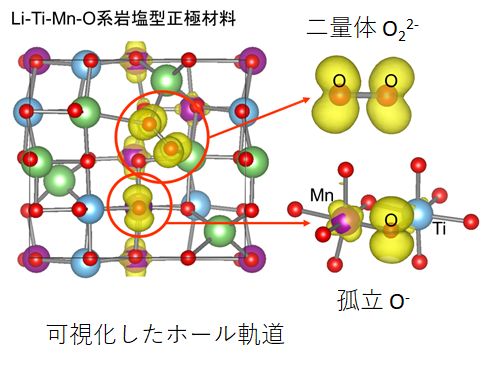
▲無秩序岩塩型構造からのLi脱離により生成するホール軌道。酸素二量体の生成や、孤立した酸化物イオンの酸化 (O-の生成)を確認した。
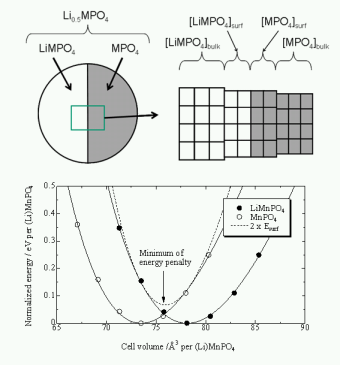
★例 オリビン型LiMPO4 (M=Mn, Fe, Co, Ni)の第一原理計算(参考文献 2008)
オリビン型LiMPO4は、いずれの中心遷移金属M(M=Mn, Fe, Co, Ni) においても、結晶構造は同一である。しかし、その電気化学特性は中心金属に対して大きく変化する。このような特性変化の理由について、包括的に理解するため、第一原理計算を用いて結晶構造・電子構造・イオン伝導・欠陥構造・界面エネルギーと電池反応との関係を定量的に調査し議論した。
★例 スピネル型酸化物LiMn2O4低温相の電子構造解析(参考文献 2010)
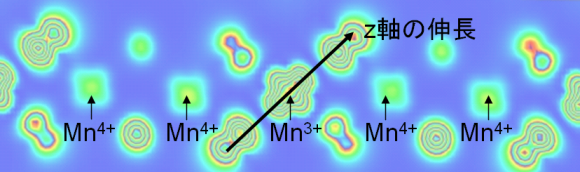
スピネル型LiMn2O4酸化物は室温より若干低い温度でMnO6八面体がひずむJahn-Teller相転移をし、立方晶(高温)から斜方晶(低温)相になる。このような構造相転移は電池材料の劣化要因や、寒冷地での機能低下になると考えられていたため、その相転移機構の解明が求められてきた。われわれは、過去に提案された巨大な超構造を構造モデルとし、強相関系の電子構造を再現するDFT+U法を適用して計算を行った。その結果、Mn3+/Mn4+の軌道・電荷整列現象を確認することに成功し、Jahn-Teller相転移(軌道整列)が電荷整列とともに発生することを明らかにすることができた。
★例 蛍石型酸化物CeO2-dにおける電子伝導(参考文献 2012)
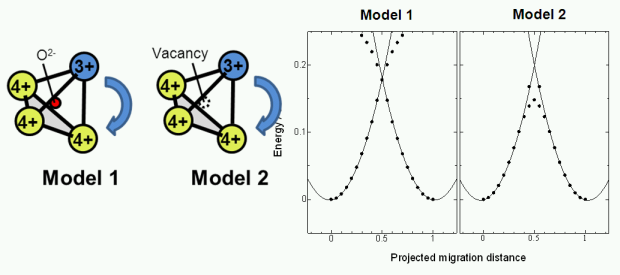
強相関系材料における電子伝導は、バンド伝導ではなくポーラロンホッピングで理解される。しかし報告される実験結果は再現性が必ずしも高いというわけではない。われわれは、DFT+U法を用いてポーラロンホッピングのエネルギーを定量的に見積もることを行っている。
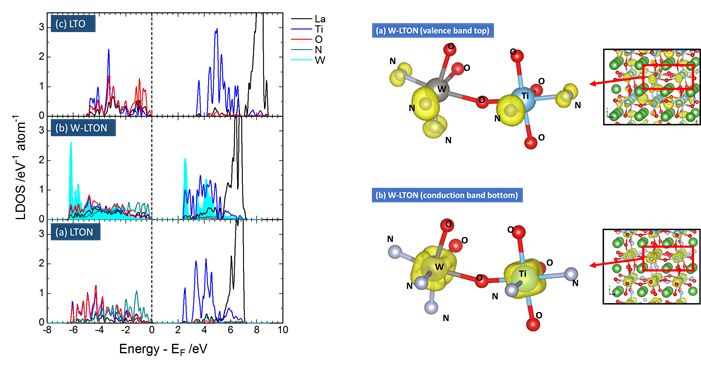
★例 酸窒化物型光触媒材料(参考文献:2016-1, 2016-2 )
光触媒として知られる酸窒化物ペロブスカイトについてドーパント導入によるバンド構造変化の詳細をバンドギャップを高精度に見積もることができるハイブリッド汎関数 (HSE06) により計算した。得られたバンドギャップは実験結果をよく再現することを確認した。(信州大学との共同研究)
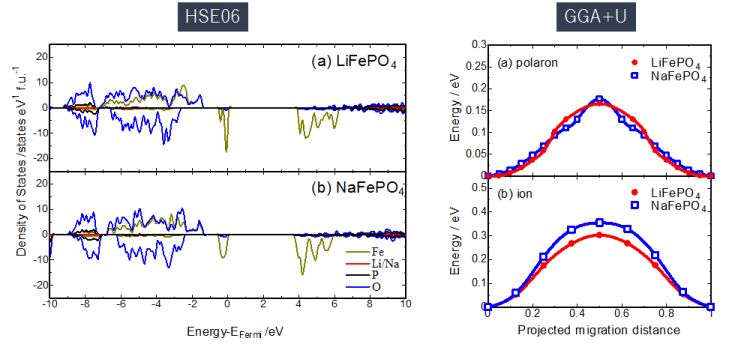
★例 オリビン型蓄電池正極材料におけるLi/Naのイオン・電子伝導機構(参考文献 2016)
LiFePO4とNaFePO4は構造も組成もほぼ等しいリチウム及びナトリウムイオン電池正極材料であるが、その電池特性は大きく異なる。その原因を調べるためにハイブリッド汎関数HSE06による高精度電子構造計算や、イオンと電子のホッピング計算などを行い、その結果を比較考察した。
★例 固体内酸素レドックス反応の解析
(6) X線吸収スペクトルの理論計算
X線吸収スペクトル(XASまたはXAFS)は、材料の電子状態を決定する有用な測定法である。しかし、スペクトルの解釈は難しく、様々な比較実験を通して類推する必要がある。われわれは多重散乱理論計算を用いて、X線吸収スペクトルを再現し、あわせて材料の電子状態を決定することを目指している。
★例 La1/3NbO3系材料の酸素 O K-edge XANESスペクトル (参考文献 2003)
オリビン型LiMPO4は、いずれの中心遷移金属M(M=Mn, Fe, Co, Ni) においても、結晶構造は同一である。しかし、その電気化学特性は中心金属に対して大きく変化する。このような特性変化の理由について、包括的に理解するため、第一原理計算を用いて結晶構造・電子構造・イオン伝導・欠陥構造・界面エネルギーと電池反応との関係を定量的に調査し議論した。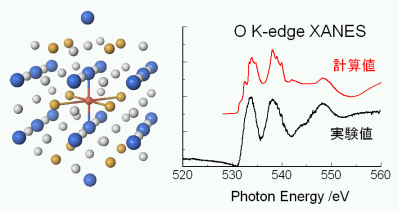
(7) 表面・界面化学反応の第一原理計算
表面あるいは界面は、バルクの材料特性とは異なる様々な機能を発現する空間である。われわれは、表面や界面で起こる化学反応を直感的・体系的に理解できるように第一原理計算を基盤とする計算技術で研究を実施しています。
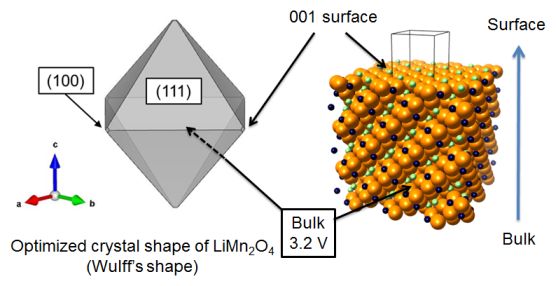
★例 リチウムイオン正極表面におけるイオン交換反応 (参考文献 2014)
リチウムイオン電池の正極材料LiMn2O4における充放電反応において、粒子表面で発生するリチウムイオンの交換反応のメカニズムを実験と計算によって検証した。優位に現れる表面を決定するためにWulffの定理を用い、さらに各表面でのリチウムイオンの交換反応速度を評価するために、リチウムイオンの表面欠陥生成エネルギーを計算している。
★例 金属表面の酸素吸着シミュレーション
金属表面での酸素吸着現象は、腐食(さびなど)や触媒特性などに密接に関連する現象である。第一原理計算により金属表面と酸素分子の相互作用を定量的に導出し、モンテカルロ法などで酸素分圧と吸着状態の関係を明らかにした。